



國產的高端新冠疫苗今(10)日進行二期臨床試驗解盲記者會,外界關注批准疫苗的緊急使用授權(EUA)上是否與美國輝瑞(Pfizer)、莫德納(Moderna)等疫苗有所差別。美國食品藥品監督管理局(FDA)早前在2020年11月訂出緊急使用授權的相關標準,疫苗解盲的審查過程採取線上直播會議,諮詢委員的投票結果也以公開具名的形式進行。
美國FDA官方網站解釋,申請EUA需要足夠的資訊證明疫苗的安全、有效性,FDA要求提交第一期、第二期的臨床試驗安全性數據,第三期的數據需要包括至少2個月的追蹤數據。FDA也要求疫苗製造商提供第三期試驗的安全性資料,必須納入至少3000人在接種疫苗後1個月後的不良反應、嚴重不良反應事件的數據。
美國疫苗藥廠要獲得FDA授權,提供的數據須包括非臨床、臨床試驗數據以及製造資訊,FDA會與藥廠事先討論第三期試驗對疫苗效力研究的預定判斷點,等到試驗進行到一定階段時,「數據安全監察委院會」獨立小組(Data and safety monitoring board, DSMB)將會對數據進行評估,根據委員的回饋報告加上FDA的意見,藥廠再決定何時向FDA提交EUA授權申請。
根據DSMB官網,該小組為國家衛生院牙科和顱面研究所(NIDSR)的獨立顧問,主要職責為:定期審查和評估累積的研究數據,以確保參與者的安全性、研究進行和進展,並在適當時評估有效性,向NIDSR提出是否進行試驗的建議。
DSMB負責對實驗數據的安全性、研究方式、科學有效性進行完整評估。除了閉門會議,DSMB會也召開公開會議,討論研究進行上的問題以及進展,開放FDA官員、研究員、行業代表等參與,但對數據的審查和審議必須保密,只有DSMB的投票成員才能取得按組劃分的結果數據的中期分析,且為確保公正性,DSMB投票成員皆不能參與評審的研究過程。

▲美國FDA對藥廠申請EUA有相當嚴格的安全標準。(圖/美聯社/達志影像)
諮詢委員會議結束後,FDA將考量委員意見,確認疫苗的可用性和安全性後決定是否批准EUA。疫苗獲得FDA的EUA批准後,還要交由傳染病防治諮詢會預防接種組(Advisory Committee on Immunization Practices, ACIP)決定相關接種政策。
根據FDA官網,目前諮詢委員會有18個成員,1個席次從缺,主席為德州休斯頓大學醫學院博士、傳染病學教授沙利(Hana El Sahly),成員包括臨床研究、兒科、免疫與呼吸疾病、病理學等各領域的醫學專家,以及行業代表「Head of Medical Affairs」副總裁、FDA生物製品評估研究中心負責人、聯邦政府指定科學顧問。
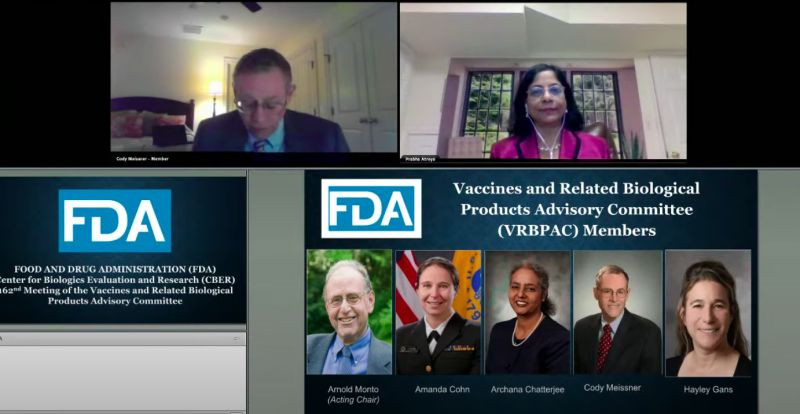
▲FDA疫苗與生物相關的諮詢委員會成員。(圖/翻攝自影片) 
▲FDA在VRPAC諮詢會議後發表聲明,FDA表示將盡快進行有關EUA的工作,同時通知CDC、「曲速行動」準備疫苗分配。(圖/翻攝自FDA)
新冠肺炎疫情爆發後不到一年,多款應用不同技術的新冠疫苗問世,大眾多少對疫苗的安全性感到憂慮。FDA強調,美國政府聯合公部門、國際社會、學術界、非營利組織和製藥公司制定全面協調策略,優先考慮發展最有前途的疫苗,聯邦政府也對疫苗產能進行風險投資,讓藥廠能夠大膽的進行開發。
輝瑞(Pfizer)-BioNTech於2020年11月20日向FDA提出EUA申請,提供的資訊包括11月對36621位參與者的雙盲測試數據,明列使用每劑使用量、2劑施打間隔的指南、疫苗保存條件、適用年齡以及可能產生的副作用與後遺症;試驗數據分析顯示,接種輝瑞疫苗可產生95%的保護力。至於安全性數據包括約3.8萬位16歲以上的參與者數據,這些人被評分為施打疫苗組/施打安慰劑組,研究人員根據FDA標準,對3.8萬人進行2個月的追蹤,發現疫苗不會造成明顯安全憂慮。
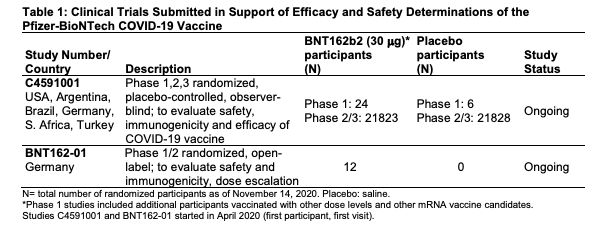
▲輝瑞申請EUA時提供的相關圖表,包括盲測數據和進行狀態。(圖/翻攝自FDA) 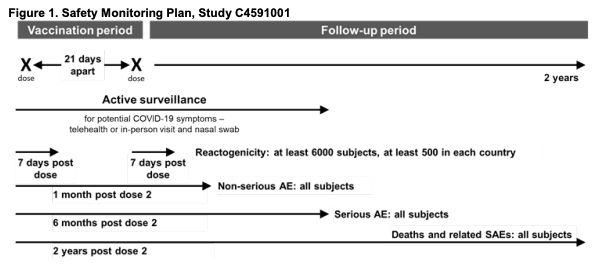
▲輝瑞疫苗申請EUA提供的安全檢測計畫表。(圖/翻攝自FDA)
美國政府根據《國家兒童疫苗傷害法》,要求輝瑞必須要向疫苗不良事件報告系統(VAERS)回報:疫苗接種錯誤,是否會造成不良反應事件;嚴重不良事件(與疫苗接種無關);兒童和成人多系統炎症綜合徵案例;導致住院或死亡的新冠肺炎案例。
疫苗與生物相關的諮詢委員會2020年12月10日根據輝瑞公司提供的數據,包括第三期試驗效用與安全性資料,進行公開投票,以17票比4票通過。考量諮詢委員會的建議,FDA隔日(12月11日)批准緊急使用授權基於試驗數據及建議,開放16歲以上人口接種疫苗,輝瑞成為美國第一支獲得EUA的疫苗。
根據輝瑞官網,在申請EUA時,「BNT162b2」新冠疫苗已經進入第三期試驗,第三期試驗已招募了超過4.4萬名參與者,其中絕大多數人已接種第二劑疫苗,分佈在美國、德國、土耳其、南非、巴西、阿根廷等150個臨床試驗地點。

▲FDA疫苗與生物相關的諮詢委員會成員公開資料(部分)。(圖/翻攝自FDA)
※【NOWnews 今日新聞】提醒您:
因應新冠肺炎疫情,疾管署持續加強疫情監測與邊境管制措施,國外入境後如有發燒、咳嗽等不適症狀,請撥打「1922」專線,或「0800-001922」,並依指示配戴口罩儘速就醫,同時主動告知醫師旅遊史及接觸史,以利及時診斷及通報。
我是廣告 請繼續往下閱讀
美國疫苗藥廠要獲得FDA授權,提供的數據須包括非臨床、臨床試驗數據以及製造資訊,FDA會與藥廠事先討論第三期試驗對疫苗效力研究的預定判斷點,等到試驗進行到一定階段時,「數據安全監察委院會」獨立小組(Data and safety monitoring board, DSMB)將會對數據進行評估,根據委員的回饋報告加上FDA的意見,藥廠再決定何時向FDA提交EUA授權申請。
根據DSMB官網,該小組為國家衛生院牙科和顱面研究所(NIDSR)的獨立顧問,主要職責為:定期審查和評估累積的研究數據,以確保參與者的安全性、研究進行和進展,並在適當時評估有效性,向NIDSR提出是否進行試驗的建議。
DSMB負責對實驗數據的安全性、研究方式、科學有效性進行完整評估。除了閉門會議,DSMB會也召開公開會議,討論研究進行上的問題以及進展,開放FDA官員、研究員、行業代表等參與,但對數據的審查和審議必須保密,只有DSMB的投票成員才能取得按組劃分的結果數據的中期分析,且為確保公正性,DSMB投票成員皆不能參與評審的研究過程。
諮詢委員會議結束後,FDA將考量委員意見,確認疫苗的可用性和安全性後決定是否批准EUA。疫苗獲得FDA的EUA批准後,還要交由傳染病防治諮詢會預防接種組(Advisory Committee on Immunization Practices, ACIP)決定相關接種政策。
根據FDA官網,目前諮詢委員會有18個成員,1個席次從缺,主席為德州休斯頓大學醫學院博士、傳染病學教授沙利(Hana El Sahly),成員包括臨床研究、兒科、免疫與呼吸疾病、病理學等各領域的醫學專家,以及行業代表「Head of Medical Affairs」副總裁、FDA生物製品評估研究中心負責人、聯邦政府指定科學顧問。
新冠肺炎疫情爆發後不到一年,多款應用不同技術的新冠疫苗問世,大眾多少對疫苗的安全性感到憂慮。FDA強調,美國政府聯合公部門、國際社會、學術界、非營利組織和製藥公司制定全面協調策略,優先考慮發展最有前途的疫苗,聯邦政府也對疫苗產能進行風險投資,讓藥廠能夠大膽的進行開發。
輝瑞(Pfizer)-BioNTech於2020年11月20日向FDA提出EUA申請,提供的資訊包括11月對36621位參與者的雙盲測試數據,明列使用每劑使用量、2劑施打間隔的指南、疫苗保存條件、適用年齡以及可能產生的副作用與後遺症;試驗數據分析顯示,接種輝瑞疫苗可產生95%的保護力。至於安全性數據包括約3.8萬位16歲以上的參與者數據,這些人被評分為施打疫苗組/施打安慰劑組,研究人員根據FDA標準,對3.8萬人進行2個月的追蹤,發現疫苗不會造成明顯安全憂慮。
美國政府根據《國家兒童疫苗傷害法》,要求輝瑞必須要向疫苗不良事件報告系統(VAERS)回報:疫苗接種錯誤,是否會造成不良反應事件;嚴重不良事件(與疫苗接種無關);兒童和成人多系統炎症綜合徵案例;導致住院或死亡的新冠肺炎案例。
疫苗與生物相關的諮詢委員會2020年12月10日根據輝瑞公司提供的數據,包括第三期試驗效用與安全性資料,進行公開投票,以17票比4票通過。考量諮詢委員會的建議,FDA隔日(12月11日)批准緊急使用授權基於試驗數據及建議,開放16歲以上人口接種疫苗,輝瑞成為美國第一支獲得EUA的疫苗。
根據輝瑞官網,在申請EUA時,「BNT162b2」新冠疫苗已經進入第三期試驗,第三期試驗已招募了超過4.4萬名參與者,其中絕大多數人已接種第二劑疫苗,分佈在美國、德國、土耳其、南非、巴西、阿根廷等150個臨床試驗地點。
※【NOWnews 今日新聞】提醒您:
因應新冠肺炎疫情,疾管署持續加強疫情監測與邊境管制措施,國外入境後如有發燒、咳嗽等不適症狀,請撥打「1922」專線,或「0800-001922」,並依指示配戴口罩儘速就醫,同時主動告知醫師旅遊史及接觸史,以利及時診斷及通報。